Dept of Psychological Medicine
Hergest Unit
Ysbyty Gwynedd
BANGOR
Gwynedd LL57 2PWTel : (01248) 384452
Fax : (01248) 371397
Sarah Wark
Medicines & Healthcare products Regulatory Agency
Market Towers
1 Nine Elms Lane
London SW8 5NQ
5th November 2004
Dear Sarah Wark
Many thanks for your letter. Given that it is roughly 5 years since I started writing to MHRA on these matters, I am sure a lot of us will be very relieved if this exercise puts the issues surrounding the SSRIs to rest.
Many thanks also for your assurances of confidentiality regarding material that I might send. I list below some material that I hope will be kept confidential. It is rather difficult however to accept MHRA assurances of confidentiality given the gross conflicts of interest between some of the experts in CSM / MCA in the past, as have been outlined recently in the media, and given the fact that if a SSRI company wanted to block publication of an article by me on say the hazards of suicide on SSRIs the list of editors they might care to e-mail would come pretty close to constituting an SSRI Review Committee, such as the one that was put in place in the first instance by MHRA. You will forgive me therefore if I find it difficult to respond uninhibitedly to your offer.
Your subsequent correspondence has also made it clear that you can do nothing to lift the constraints of confidentiality orders that I remain under, and you will need to bear this in mind when interpreting my response.
I take it from your letter that you will bring the material outlined here to the attention of the SSRI review committee, but I would be obliged if you can confirm that this letter has in fact been given to all members of the committee.
Regarding the material you have asked for, I enclose the following:
1/ The meta-analysis you asked for – this is marked appendix 1. This is at present unpublished, so I would appreciate if you could treat it confidentially.
2/ A review on suicide on antidepressants that you may be unaware of, authored by Graham Aldred and myself. This is also at present in press and I would appreciate if you could keep the contents confidential for the moment. This is marked appendix 2.
3/ A further study, to whose existence I alerted Dr Raine some months ago – this is marked appendix 3. This appears to remain at present unpublished, and therefore again confidentiality would be appreciated.
I wish at this point to draw your attention to a number of other datasets.
4/ In a 50 page communication posted on the FDA website in connection with the recent paediatric advisory committee on antidepressants, Pfizer offered two further sets of figures that should be of interest to the review body. This letter, referred to below as Ryder (2004), can be accessed on:
www.FDA.gov/ohms/dockets/ac/04/briefing/2004-406561-31-pfizer-letter.pdf
Anyone from the review committee reading this though will need to bear in mind that, although apparently a scientific piece, this is not written by a scientist, but rather by a lawyer, Judith Lonsdale as I understand it.
Sertraline Analysis 1:
First on page 41, Ryder/Lonsdale demonstrate quite clearly that a significant number of suicidal acts in their clinical trial programme occurred in the washout/run-in phase of their trials as well as in the post continuation phase of the trials. Post continuation in this context means both several weeks after the trials were over and in trials not officially part of the NDA.
They note that FDA – and I presume MHRA – were aware of this manipulation of the figures and did not ask for any further analysis.
If we exclude all run in and post completion acts, this seems to show 1 suicidal act in 786 patients on placebo, and 7 suicidal acts in 2,053 patients on sertraline. This gives a relative risk of suicidal acts on sertraline compared to placebo of 2.69.
In fact, this misses out one suicide – the post continuation phase suicide for sertraline listed was not post-continuation. Including this would give a relative risk of 3.07 for suicidal acts on sertraline compared to placebo.
But in addition, it is no by means clear that the suicidal act on placebo was in fact such an act. Having reviewed the case narratives for all suicidal acts I think there are grounds to doubt this, which would of course give an infinitely greater relative risk of a suicidal act on sertraline compared to placebo.
Sertraline Analysis 2:
On page 24/25 in the text of their letter to FDA, Pfizer claim to have undertaken an analysis in May 1992, which was restricted to placebo controlled trials. In this they claim that there were 3 suicidal acts in 926 placebo cases, a rate of 0.32%, versus 4 suicidal acts in 1424 sertraline cases, a rate of 0.28%.
This is a particularly interesting analysis for a number of reasons.
First there is the almost unbelievable manoeuvre Pfizer undertake in this document, whereby they literally compare suicidal acts with suicidal acts alone and omit all suicides, of which there were two on sertraline and none on placebo. This is either an extraordinarily Jesuitical manoeuvre, or can be explained by the fact that the Pfizer analysis for FDA, back even then, was written by a lawyer rather than a scientist.
I wish I had confidence that the data manipulation involved here might have been picked up by MHRA reviewers, however, to be fair to any reviewer, a failure to pick up the problem would possibly owe more to a reviewer being disarmed by the disbelief that such a manoeuvre might be undertaken than it would owe to anything else.
Second Pfizer do not include 2 placebo controlled studies, that later involve randomised withdrawal, in which there was 1 suicidal act on sertraline with none on placebo.
Of the three placebo suicidal acts mentioned, one was the suicidal act outlined above, for which I cannot find any evidence for a suicidal act. A second came from a UK trial, which I understand Swedish regulators panned as incapable of yielding evaluable data. The third may have been the only genuine placebo suicidal act.
By my reckoning therefore there are either 3 suicidal acts in 1055 placebo patients but more probably 2 suicidal acts in 1055 patients, and possibly even 1 suicidal act in approximately 1000, patients.
Against this there are 7 suicides plus suicidal acts (5 suicidal acts and 2 suicides) in 1611 patients on sertraline.
This would yield an Odds Ratio of 1.53 if there are 3 suicidal acts in the placebo group, or 2.29 if there are 2 suicidal acts in placebo group.
As of this point in time I am only in possession of the full dataset from one other placebo controlled trial on sertraline in adults, undertaken by Ulrik Malt, who found 1 suicide and 3 suicidal acts in 122 patients on sertraline, versus 0 suicides or suicidal acts in 129 patients on placebo. This study completed circa 1994 but published in the BMJ in 1999 contained no reference to suicidal acts.
Adding the Malt figures to the others above yields figures of 11 suicides and suicidal acts from 1733 patients on sertraline versus 2 suicidal acts in 1184 patients on placebo, which gives a relative risk of 3.76.
As I understand it in later documents in which suicides in sertraline trials were assembled, the figures for suicides on sertraline versus placebo may differ to the extent of 24 v 0.
The Pfizer defence of their actions in this regard is quite illuminating – basically it comes down to FDA, and, although not stated, MHRA, did not object to what we did. This seems to point the gun barrel of blame squarely at the regulators.
Sertraline Evidence of Inefficacy
It is instructive to contrast the evidence for efficacy of sertraline at roughly this time, 1994, with the evidence for risk of suicide induction, to see how the risk benefit ratio looked then, and the regulatory response to the respective datasets.
It appears that the United Kingdom was the first major country to license sertraline. It had done so before the US, where FDA officials took an active role in trying to persuade their psychopharmacological drugs advisory committee that sertraline should be licensed, even though a preponderance of the evidence did not point to evidence of efficacy.
FDA officials at the time made it clear that they did not expect sertraline to get licensed in the rest of Europe – see Leber memoranda August and December 1991 (appendices 4 & 5). Another FDA official described the sertraline portfolio of studies as embarrassing – Brecher memorandum November 1991 (appendix 6).
Basically the evidence for efficacy in 1991 was much less well established than was the evidence for suicide induction. At that point in time, only 1 of 5 studies was convincingly positive for sertraline. By 1994, when the relative risk of suicidal acts from placebo controlled trials was of the order of 3.76 for sertraline compared to placebo, only 5 of 16 sertraline placebo controlled trials, as I make it out, could be regarded as positive for efficacy for sertraline. Yet CSM/MCA had passed this drug when no other major country in Europe had done so, and nor had Australia, Canada or other countries, and CSM/MCA had licensed sertraline without warnings, and saw no reason it seems to reassess their position on this.
Against this background, and the details outlined below regarding regulatory complicity in the problem, questions have arisen regarding the influence of Dr Stuart Montgomery on CSM’s/MCA’s framing of the issues. It is worth noting that the document made public recently indicating Dr Montgomery’s links to Pfizer and advice to them regarding sertraline, none of which apparently were declared, while functioning at the same time as the psychiatric expert on the CSM, is not the only document available.
Sertraline, Paroxetine, Fluoxetine & Regulatory Complicity
In February 1990. the Teicher article raised concerns that the recently licensed fluoxetine might trigger suicidality in depressed patients. In fact, as the meta-analysis of the clinical trial literature on SSRIs mentioned above (appendix 1) and submitted to you indicates, as of 1988, there was a demonstrable doubling of the relative risk of a suicidal act on an SSRI in these trials compared to placebo, even though the data on zimelidine had not been reported in the published papers.
By October of 1990, FDA had decided the issue of suicide on antidepressants was as Martin Brecher of FDA put it: "not .. a real issue, but rather as a public relations problem" (Brecher 1990)(appendix 7). My question is whether CSM/MCA had made a similar decision.
This determination that the issue of suicide induction was a public relations rather than a substantive issue was made without holding a scientific advisory meeting. When FDA held an advisory meeting on the issue of antidepressants and suicide in September 1991, evidence on two other SSRIs, sertraline and paroxetine, already with FDA for close to two years, was withheld from the meeting. The suicide and suicidal act data on SSRIs or other antidepressants has never been shown to an FDA advisory panel and has not until this year been reviewed thoroughly by MHRA. Viewing the issue as a public relations matter has been a crucial misstep that I would argue has led both regulatory agencies down a path of twisting the science to fit their position.
The clinical trial evidence from paediatric populations conducted from the mid-1990s onwards has recently been shown to reveal that the risk ratio for suicidal acts (there were no completed suicides) on antidepressants in minors compared to placebo. But clinical trials submitted to the FDA as the basis for approval of these drugs for adults, show a risk ratio for suicidal acts on antidepressants compared to placebo of 2.39 (95% C.I 1.66 – infinity; p = 0.0001) and for completed suicides of 4.40 (95% C.I. 1.32 – infinity; p = 0.0125) (See Healy and Aldred - appendix 2). A pooled analysis of trials available as of 1991, even though these had not been designed to investigate the problem properly, revealed an approximate doubling of suicide rates.
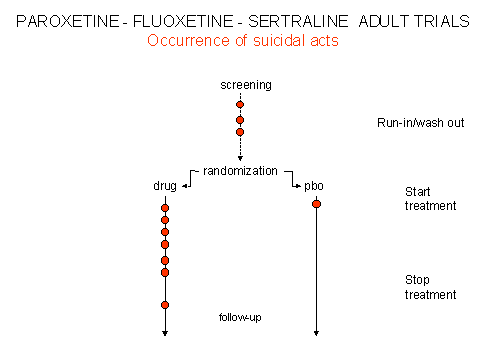
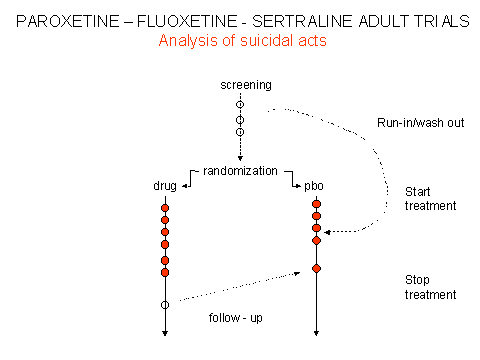
Although data submitted to FDA and CSM/MCA showed that there was an increased rate of suicides on antidepressants compared with placebo, this simple but crucial finding was obscured and continues to be obscured by two sleights of hand. First the results were biased by the inclusion in the placebo group of "washout" or "run-in" suicidal acts that had occurred before patients were randomised, as outlined above for the sertraline data. This spuriously increased the placebo rate. A further biased elevation of the placebo rate occurred by including post-continuation suicidal acts under the heading of placebo. These were acts that occurred either after patients had completed the randomised phase of the trial, and were potentially in withdrawal, or were taken from selected trials that were not part of the original clinical regulatory trial portfolio. The effect of these manipulations is laid out schematically in figures 1 & 2.
There is no record of a manipulation of this type being undertaken by any drug company for any other drug, but curiously it was done for the three major SSRI antidepressants submitted to both FDA and CSM/MCA. How is it possible that three drug companies each suddenly decided to inflate their placebo rates by including cases that were never randomised to placebo?
In the wake of the "public relations" concerns about fluoxetine, FDA invited SmithKline Beecham to withdraw their original data submission and submit a fresh set of figures for suicidal acts on paroxetine (Brecher memorandum October 1990; Appendix 7). This, the company did, in the process recoding run-in and post-continuation data as outlined in figures 1 and 2. Pfizer and Lilly did just the same. FDA monitors noted this recoding at the time. MHRA monitors appear to have noticed this in 2004 – it would be very interesting to know whether it was noted before.
Against a background of memoranda indicating that in 1990 FDA regarded the question of suicide on SSRIs as a public relations issue, and the curious decision by three companies to invoke a plainly invalid statistical manipulation, it is difficult to avoid the conclusion that the failure by the regulators to admonish the companies of this improper manipulation suggests regulatory complicity in a process intended not to inform but rather to support a position that the regulators had already decided was in the public’s best interest. Pfizer’s rationale of their involvement in this practice for data on sertraline is illuminating. Having made it clear that: "Pfizer’s 1990 report to FDA plainly shows … that 3 placebo attempts as having occurred during single blind placebo phases", they add … "FDA has neither criticized these data or the report as inappropriate, nor required additional analyses" (Ryder 2004).
If companies manipulate data like this and regulators miss it, a good argument can be made that the companies have done nothing illegal – even if the practice breaches norms. A sharp practice like this perhaps fits understandings that companies will try to present their data in the best possible light. However, if the regulators have been more actively involved in the process, the outcome is more serious, precisely because no-one expects this of regulators.
A second problem is that the data were inappropriately subjected to tests of statistical significance even though it was known in advance that the studies were so small that if an individual antidepressant doubled the risk of suicide, an increased risk of this magnitude in these studies could not possibly be statistically significant. Which is exactly what happened. But instead of reporting that there was an increased risk of suicides for people randomised to antidepressants, it was widely reported that there is no statistical difference and the evidence of increased risk was not "credible". Such claims continue to be made, not least by MHRA/CSM officials.
This approach to data on safety is asymmetric to the approach taken by FDA and MHRA on efficacy, as outlined above for sertraline, where not all trial failures to beat placebo are counted. Trials are seen as "assay systems" and any positive results greatly outweigh what may be an overwhelming majority of negative results. In the case of sertraline, this has meant that, where only 1 of the initial 5 trials produced a clearly positive result and only 5 of the first 16 trials were clearly positive, such that there were concerns as to whether this drug should be licensed, FDA and MHRA opted to be guided by the signal of possible efficacy that came from a small subset of trials. Sertraline is now the best-selling SSRI in the world, marketed under the banner of unparalleled evidence of efficacy.
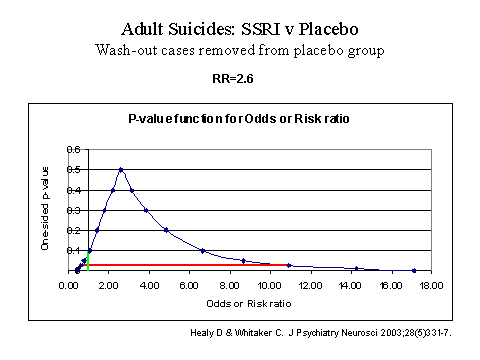
When it comes to safety, the regulators could now argue that as one assay system, the paediatric trials, have established a causal link between antidepressants and suicidality on the basis of pooled data, leading to warnings for children being put on antidepressants in the United States, as of October 2004, warnings should be more generally attached to the drugs. However, instead, MHRA appears to continue to insist on an all but unreachable threshold. For individual drugs, where a confidence interval includes 1.0, as in figure 3 below, MHRA have stated repeatedly that a failure to reach statistical significance means that there is no credible signal of a suicide risk. These statements have been made even though these trials were not powered to test for suicide risk, and significance testing should not be applied. Confidence intervals were designed in fact to avoid premature rejection of possible linkages. In this case, the correct interpretation of data such as those in figure 3 is that the best estimate for likely risk of suicide on SSRIs over placebo is 2.6 and the scientific data is consistent with a possible 10-fold increase in risk. I wonder whether this inappropriate turn to concepts of statistical significance stems from the need for regulators to make decisions and a naïve hope that a simple decision making rule will take the risk of decision-making out of their hands.
There is a third problem with the data from these trials. In the case of sertraline, and possibly also in the case of paroxetine and fluoxetine, FDA officials appear to have taken a further initiative to suggest to Pfizer that they undertake survival analyses of their data, and in particular that they should control for exposure to the drug in a manner that assumed there was a constant hazard from treatment rather than particular risk periods. Survival analyses are an entirely appropriate method of data handling, which are easier if the hazard is thought to be constant, but the clinical literature had pointed strongly at this stage to clear risk periods rather than a constant hazard. The assumption of a constant hazard, which averages out periods of greatest risk, was inappropriate.
The scientific literature on these issues since 1990 has presented the data as outlined in figure 2 rather than the data in figure 1. Combining both the recoding approaches and survival analyses outlined here in a paper on suicidal acts on paroxetine, Stuart Montgomery, who was the psychiatric expert on the CSM during this period, claimed that the data on paroxetine showed a 5-fold greater risk of a suicidal act on placebo compared to paroxetine, when in fact the raw data point to a relative risk of suicidal acts of 4 or greater on paroxetine compared to placebo (Montgomery et al 1995). Dr Montgomery it would seem since claims that he never got to see the raw data behind this article, even though the data are the data submitted as part of the application for a license for paroxetine, which he was party to recommending.
This and other analyses of individual drugs (Beasley et al 1991), and multiple analyses of drug groups (Khan et al 2000 & 2003) have followed figure 2, even though the true disposition of the data, as in figure 1, has been in public domain for a decade previously in the form of FDA medical and statistical reviews of these drugs.
Despite recent warnings on antidepressants for children, regulatory embroilment in these issues continues. An FDA analysis of the data for suicides from adult placebo controlled trials of antidepressants published in a brief abstract indicated that the risk of suicide on treatment is approximately double in adults (Hammad et al 2003). But there are two puzzling aspects to the presentation of these data. First FDA claim that by controlling for an unspecified set of factors they can eliminate this apparent risk, without indicating how this could happen, and anyway it is not clear that manipulating data from randomised controlled trials in this post-hoc way is appropriate, given that randomisation is explicitly designed to control for confounding. Second, the FDA officials responsible for this analysis refuse to hand over the data for confirmatory analyses or to otherwise put the data in the public domain. This arguably breaches the norms of science, and is certainly inconsistent with what both FDA and MHRA have recently done with data on suicidal acts in paediatric trials. Under scrutiny from Congress, FDA claim they need to reanalyse data that they have had for over a decade. MHRA as both know have been analysing the data for the better part of two years.
FIGURE 1
FIGURE 2
FIGURE 3
Withdrawal
In the SSRI expert working groups’ review of the data on Seroxat I understand they will have been faced with presentations of the data on withdrawal from Seroxat by the market authorisation holders. These presentations to judge by material I have seen have made it clear that the company has excluded a large number of reactions from further analysis, owing to a prior determination that these reactions are not caused by withdrawal.
Such reactions include respiratory disorder, infection, depression, emotional lability and abnormal ejaculation for instance. Removing these from any calculation in my estimation would remove approximately 20% of withdrawal events.
MHRA reviewers have made no objection as I understand it to such a priori exclusions, but on simple methodological grounds a failure to object is extraordinary. If MHRA reviewers have in fact failed to object, I suggest the MHRA revisit this issue in the light of the points made below.
In the clinical trials of all SSRIs, respiratory infections and pharyngitis are recorded as occurring at a much greater rate than on placebo. When the relevant studies, such as studies on healthy volunteers for instance, have been conducted by clinicians with expertise in these areas, it has become clear that these respiratory infection and pharyngitis reactions in many cases are dystonias and dyskinesias of the jaw, throat and upper respiratory system rather than infections. The occurrence of such effects on withdrawal have also been reported and indeed would be expected if such reactions are triggered by the initial exposure to treatment.
I think your reviewers have been fooled by a coding manoeuvre here.
My expectation is that Glaxo SmithKline will also have removed from the category of possible withdrawal reactions, patients suffering from emotional lability, when it is now clear that such patients were most probably suicidal, and as of June 2003 even Glaxo SmithKline have written round to physicians in the UK conceding that emotional lability is linked to suicidality and to withdrawal. Despite this, I suspect there have been no objections by MHRA reviewers in the course of 2004 to Glaxo’s removal of such data from their consideration of the rate of withdrawal reactions following cessation of treatment with Seroxat.
If there have been no objections, this would appear to be a case of MHRA reviewers failing to join up the dots.
I would note furthermore that the occurrence of emotional lability/ suicidality in the withdrawal periods of trials across indications, including for instance OCD and social phobia studies, argues strongly against these phenomena being linked to the disease and strongly in favour of a linkage to treatment.
I have also seen Glaxo SmithKline remove abnormal ejaculation from the list of possible withdrawal reactions. This is quite extraordinary as the effects of withdrawal on ejaculation and orgasm have been well described for 20 years for tricyclic agents, such as clomipramine, which have effects on the serotonin system. It is difficult to believe that MHRA would not protest at the exclusion of such effects.
An unquestioning acceptance of these exclusions however adds to a consistent pattern of MHRA reviewers taking on trust company assessments of their own data. This is a pattern that may unfortunately make it difficult for many to have confidence in MHRA’s final report.
On the same theme, MHRA do not appear to have questioned Glaxo SmithKline’s suggestion that hostility (aggression) in their healthy volunteers taking Seroxat stemmed from the volunteers being cooped up. This highly convenient excuse seems to have been swallowed uncritically by MHRA, even though evidence from trials of Seroxat in children, as well as Seroxat in PMDD, point strongly to a hostility inducing effect of the drug.
Duloxetine
Although you have recently licensed duloxetine for use in woman with no mental health problems and have done so without warnings, it seems an idea to make sure you have the following data.
First, my understanding is that the relative risk of a suicidal act from the duloxetine clinical trials in depression approaches double the rate found on placebo, and the scientific data are consistent with a 4-fold increase in the risk of suicidal acts.
Second, the rate of suicidal acts in duloxetine open trials for stress induced urinary incontinence is at present 14 times the expected rate from the normal population.
Finally the clinical record prior to her death of the healthy volunteer Traci Johnston who had just begun withdrawal from a supra-therapeutic dose of duloxetine shows a striking overlap of symptoms with those recognised to form part of a withdrawal syndrome for venlafaxine, which of course is now clearly linked to induced suicidality.
GPRD Studies
In recent contacts with the media it would seem that MHRA have put store on an analysis of GPRD undertaken by Dr Herschel Jick and colleagues. MHRA appear to be still briefing the media that this study shows there is no problem with antidepressants, even though FDA have put alternate figures from this study into the public domain.
You will know that Jick et al (2004) reported that when dothiepin was used as the reference compound there was a considerable difference between these agents, although at a 95% confidence interval there was not a clear separation between dothiepin and the SSRIs, fluoxetine and paroxetine. However, when amitriptyline was used as the reference compound, the figures for dothiepin were 1.21 (95% C.I 0.89 – 1.64), for fluoxetine 1.46 (95% C.I 1.03 – 1.91), and for paroxetine 1.55 (95% 1.11 – 2.16) (FDA 2004). These figures were presented at the FDA hearings of September 13th and 14th, which I am told MHRA were following closely.
That the results would come out this way seemed highly likely from a simple consideration of the confidence intervals in the published study. It would seem that Dr Jick chose to publish the only possible analysis of his data that would not incriminate the SSRI’s unduly.
As regards, the further findings from the Jick study indicating that the risk of suicide was 38 times greater in the first 9 days of treatment, the notion that this might stem from the disease rather than from the treatment is comprehensively refuted by the general clinical trial data for SSRIs undertaken in adults. This is particularly so, given the difficulties the SSRIs had in distinguishing themselves from placebo.
The argument has also been put forward that perhaps more severely depressed patients were preferentially given SSRIs and that this confound might explain the results found in the Jick study. But this too is unlikely in the light of the unpublished Montgomery data on fluoxetine and paroxetine, which controls for confounding and indicates a treatment-induced exacerbation of risk in patients prone to suicidal acts.
I find it astonishing that MHRA would refer to the Jick study as evidence that the drugs do not cause a problem. I also find it astonishing that Glaxo SmithKline at least and possibly also Pfizer have had access to GPRD, while a study MHRA was supposed to have undertaken on GPRD, the results from which as I understood it were to have been made public last April, have not be reported on.
You will appreciate that, as Dr Jick appears to have shown, wittingly or not, given enough time it is always possible to find ways to cut GPRD data in a manner that may appear to minimize the problem. Allowing the companies to prepare a set of defenses without making possibly already available data open for scrutiny smacks of an extraordinary one-sidedness.
Prozac and SSRIs for Children
Since I attended the SSRI review committee, the question of the use of Prozac in children has come to the fore. Prozac is apparently under review for children by MHRA at present and the impression stemming from correspondence that is in the public domain from Lilly and from other regulators in Europe is that approval by MHRA is interestingly all but certain.
I have had a chance to review a good deal of data on Prozac given to children and wish to make the following comments to the committee, many of which will not come as any surprise to you.
There have been four trials of Prozac in children who are depressed. The first by Simeon et al 1990 showed no differentiation between Prozac and placebo.
The second by Emslie published in 1997 showed no differentiation between Prozac and placebo on the primary outcome measure of the study. This was a trial in which less than 100 children were selected from over 500 children screened. This trial also used a placebo washout period to exclude placebo responders.
The third trial was a most unusual trial. Emslie was again the first author, and this trial was published in 2002. In addition to extensive screening to select patients likely to be responsive to treatment, this trial had a placebo washout phase to exclude placebo responders, as had the previous Prozac trials, but in addition it had a further phase, which involved children randomised into Prozac arm of the trial being given a week of Prozac 10mg first with an exclusion option for any children who appeared to respond adversely to Prozac. The trial proper started after this extraordinary manoeuvre.
Even with this extraordinary step on the primary outcome measure in the trial as indicated in FDA medical reviews of all of these trials Prozac did not differentiate from placebo.
The fourth trial you’ll be aware of has recently been published in JAMA with John March as a first author, having first featured in the New York Times and elsewhere extolling the virtues of Prozac. I think rarely will the abstract of a major paper have been so at odds with the underlying raw data.
I understand some of you in MHRA were watching the feed from the FDA paediatric advisory committee meeting. If that is the case presumably you will have seen Dr March present the data there and heard him questioned. One of the most striking features of this interchange was that Dr March was forced to admit that according to strict FDA criteria that Prozac in his trial had not been shown to work either.
The March group had manipulated the data in an interesting fashion, which involved searching for responders and non-responders on scales such as the CDRS or the clinical global impression scales, and compared these. When this is done, the effect is to squeeze patients into categories and under those circumstances Prozac can be shown to be different from placebo but this squeezes the middle of the clinical population in a manner that is methodologically inappropriate. When the CDRS or other scales are used on a continuous variable basis, as per FDA requirements for demonstrating efficacy, the differences between Prozac and placebo are not statistically significant. This point was recently brought out in a position piece by Newman in the NEJM.
You will no doubt also be aware that the rate of suicidal acts on Prozac in this latter trial is no less than the rate of suicidal acts on other SSRIs, and is in fact substantially greater than in most other SSRI trials.
It would seem therefore that there is no more evidence of efficacy or for safety on Prozac than there is for other SSRIs, and just as much evidence for harm.
However it is also worth noting that there is some evidence for efficacy for Prozac, Seroxat/Paxil and Zoloft/Lustral for OCD, but at present it is difficult for any clinician to use an SSRI for any child or teenager in the UK, owing to MHRA’s handling of the issues thus far.
For the record it’s perhaps worth adding at this point that I don’t agree that MHRA made the correct move in contraindicating SSRIs in children. There is some evidence from trials in OCD and other anxiety disorders that some of these drugs can be effective. The key issue is that the treatment should come with the proper warnings for both adult and paediatric populations.
My suspicion is that, as MHRA rarely if ever do anything without some consultation with the market authorisation holders, it appeared to be a better bet to the market authorisation holders to have the drugs contraindicated in children, given that so few children in the UK were having these drugs, rather than to have appropriate warnings placed on the treatment, as such warnings might impact on the adult market. But contra-indicating SSRIs and other antidepressants is the move that seems to have set up the current quandary MHRA are in, which appears to have all but required a licensing of Prozac for children.
If Prozac is licensed for children, it is also worth noting that there are other features of Prozac use in children that are of concern. The FDA reviews of this drug make it clear that children taking Prozac failed to grow at the rate that children taking placebo grew. The FDA asked Lilly to undertake follow-up studies of this effect, but as far as I know the company have not done so. No doubt MHRA will have noticed this problem in the trial data also. If you do intend to license Prozac, it would be good to know that the issue of growth has been investigated properly.
There is a further potential hazard of Prozac that FDA could not have been aware of when they reviewed the drug, but which MHRA have a chance to familiarise themselves with. In April of this year the US Department of Health and Human Services, National Toxicology Programme, issued a Centre for the Evaluation of Risks to Human Reproduction Report that focused on Fluoxetine – NTP CERHR Expert Panel Report on the reproductive and developmental toxicity of Fluoxetine. In this it is made clear that in male animals tested Prozac leads to testicular shrinkage of approximately 25%. The use of this drug in men generally must therefore be of some concern. The use of this drug in pubertal boys would seem distinctly problematic.
Aggression and Hostility
On the issue of aggression and hostility, you have asked about the origin of the figures quoted in the Guardian in September 21st, in the light of a discrepancy between my analysis and what you suggest you have found.
One source was Glaxo SmithKline’s trials. Even before posting edited summaries of their trials, in June this year, they had posted the following:
Glaxo SmithKline (2003). Data on File. Important Safety Information regarding Paxil in Pediatric Patients, Glaxo SmithKline, Therapeutic Products Directorate: TDP-Web, July 18th 2003. Health Canada, www.hc-sc.gc.ca/hpfb-dgpsa/tpd-dpt/paxil_pa_e.html
But you will appreciate also that I have seen a great number of the healthy volunteer studies SmithKline undertook.
And in the case of the children’s data, I have had access it would seem to a greater number of such studies than you had – at least until comparatively recently. One of these studies, 716, for instance, was placebo controlled and had an OCD arm to it, and it seems unlikely to me that you had been including this in your analyses to date.
But in addition to the above data on paroxetine and the light these shed on the issue of SSRIs and hostility/aggression, I would like to draw your attention to the following material on sertraline.
As of 1996, in conformity with requirements for standard International Product Information for Sertraline, Pfizer in the UK harmonised the labelling of Sertraline to show the following effects – depressive symptoms, agitation, anxiety and psychosis, in addition to leaving there aggression and hallucinations. Aggression in this context is a coding term that includes homicide.
A study of the Summary of Product Characteristics, I believe makes it clear that these events were considered related to sertraline and do not fall into the sub-categories of events where it was unclear whether sertraline had a causative role. Determinations to include effects such as aggression and to link them to sertraline in this way will not have been made without a clear review of the data within the company.
While material to this effect is included in the product labelling of sertraline in the UK, there would appear to be compelling grounds to alert clinicians more explicitly to the possibilities of psychosis and aggression, as you cannot depend on the company or companies to do so, and you cannot depend on clinicians to read the small print of product labelling or understand distinctions between some of the ways events are categorised in the labelling. There has also of course been a sustained campaign by all of the SSRI companies to deny that anything like this could happen on their drugs.
I accept that attempting to reverse a decade of misinformation puts MHRA into difficult waters, in that it is not your job to ensure that the average clinician either reads or fully understands the nuances of product labelling but as recent reports have made clear, reactions of this sort are happening, and given the volume of prescribing such reactions are not infrequent in absolute numbers. Clinicians here in Britain, however, regularly respond when faced with such problems by increasing the dose of treatment rather than by considering the possibility that treatment may be the causative factor.
As the current product labelling stands and against a background of bland reassurance, most clinicians will not read the labelling to mean that a causal link between sertraline and adverse reactions such as psychosis and aggression has been established.
Correspondence with MHRA
I have never had formal feedback from MHRA on either of my two oral submissions to you or on the documentation subsequently sent to you in terms of the adequacy or otherwise of the material presented or sent. I have had informal feedback, however, that has suggested that I am viewed within MHRA as having been in some respects unhelpful when approached on issues.
The only issue that I believe I’ve been approached on specifically, other than the Prozac data you requested, outlined below, had to do with a clinical trial undertaken by Stuart Montgomery. I was asked whether I could identify the protocol number of a trial undertaken by Dr Montgomery in recurrent brief depressive disorders. Given that at the time of this request, you had Dr David Baldwin as a member of your review committee who was an investigator in that same trial, it seemed to me extraordinary that you had to turn to me for an answer on this point.
It also seemed odd to me afterwards that anyone should hint at me being difficult on the matter of identifying protocol numbers when I remained – and remain - under confidentiality orders regarding the same material, and I thought I had made this abundantly clear. You also have now confirmed that you either cannot or will not subpoena me.
I have however since had a chance to review all of the protocol numbers of Glaxo SmithKline’s trials as submitted to you. This led me at one point to consider that protocol 57 might represent the Montgomery study. But I now suspect that the Montgomery study may not be represented in any of the protocols you have.
There is every reason to believe that you still do not have the data on this important study. I think I now understand how this could arise. Dr Montgomery initiated a number of studies himself. These studies may have been funded by GlaxoSmithKline, or other companies, subsequently, or in indirect ways. Either way, the net result is that the studies with this particular patient group may have taken place outside of the usual company methods of organising clinical trials.
It is quite possible therefore that perhaps the only study ever formally designed to look at the effects of paroxetine on suicidal acts is one that has still not been submitted to you by GlaxoSmithKline but that they have not broken any laws in not submitting it to you.
I am clear however that you should be able to get hold of this study from GlaxoSmithKline, as I am certain the company have a copy of the full details of the study. Alternatively Drs Montgomery or Baldwin, both of whom have been experts in CSM or in MHRA’s SSRI expert review groups, should be able to afford you a full set of details.
I would appreciate if you could confirm for me that in fact you do have this study. The fact that such studies might exist outside the usual framework, and that MHRA might not be able to access them is quite a worrying prospect.
I would also appreciate clarification as to whether you still think I am being uncooperative in the production or non-production of identifying details for this study. Ideally it would be helpful to get a comment as to whether you find my surprise that you should turn to me for these details, rather than to Drs Montgomery or Baldwin, unreasonable.
You might also care to note that there is a similar study undertaken by Dr Montgomery on Fluoxetine that Lilly may not have submitted to you in which placebo beat Fluoxetine at a significance level of P=0.006. This would seem to be a study that MHRA should get their hands on, if they do not yet have it.
Prozac in Adult Studies
Under this heading you have asked the following questions:
Q. Confirmation that the data provided are from studies HCAA, HCAB, HCAC, HCAD, HCAF and HCCG.
A. I believe this is correct.Q. Copies of protocols for each of the above mentioned studies.
A. I do not have the protocols for each of these studies to hand. I find it astonishing that you have not sought the protocols from the market authorisation holder.It is approximately two and half years since you suggested sending this material to you. The administrator who did all the copying has since left, as indeed has her successor. The files are all boxed and stored, as I have had no occasion to work on Prozac since – unlike MHRA I am not reviewing Prozac for any purpose.
Furthermore, at present I unfortunately do not have administrative support and therefore cannot undertake to find this material within a week, which is the timeframe that you have given me.
Q Confirmation of the number of patients in the placebo and active groups for each study.
&
Q Confirmation that we have received the complete set of patient records for each of the studies.
A The common answer to these questions is that, as I understand it, Lilly did not provide plaintiffs with a complete set of records for these studies, even though the plaintiffs’ lawyer in question, Nancy Zettler, understood that they had done so. It was only when the plaintiffs expert in Fentress v Eli Lilly, Dr Lord, sought to offer a view on the records, and Lilly objected on the basis that she had not seen a complete set of records that it became clear that the records you have here may not be a complete set.
The person who can clarify this point is Nancy Zettler, who can be contacted on Zet2400@aol.com.
Q Clarification on how you consider these data will inform the ongoing work, in particular which aspects best illustrate your concerns about
i) Miscoding of suicidal acts as failure to respond to treatment rather than adverse effects;
A It is clear that if you study the records on Prozac, Seroxat/Paxil and Lustral/Zoloft as I have that the companies do not have a good idea of what happened in their own trials. Reports on these trials list patients who have committed suicide, and list those patients as being of a certain age and as having committed suicide at a certain point during the trial, when the patient in question has a very different age and the event in question happened at a completely different point during the trial.
A significant number of those deemed treatment non-responders by the company in the Prozac trials you have will I believe on inspection turn out to be cases of developing agitation, suicidality, confusion, psychosis and other mental decompensations that are at least as likely to be a consequence of Prozac as they are to have stemmed from the disease being treated.
ii) Miscoding of suicidal act as emotional lability;
A The issue of miscoding suicidal acts as emotional lability is not one I believe that I introduced at the point I offered to send you the data on Prozac. Emotional lability has been a Glaxo SmithKline speciality. Lilly have resorted to treatment non-response and a range of other headings to code what happened.
Basically if you look in detail through the records on Prozac, Seroxat/Paxil and Lustral/Zoloft, you will find cases of homicidality coded as nausea for instance. A relatively quick interrogation of the records from Pfizer’s early Lustral/Zoloft trials should show a number of patients with inappropriate codings like this.
Depending on what clarification you can shed on the issue of confidentiality, I can identify specific trials for sertraline and paroxetine and patients that fit just the issues on which you appear to want answers, but otherwise I remain constrained by confidentiality orders.
iii) Discontinuation of patients from studies for primary adverse effects such as nausea when in fact there has been a suicidal act;
&
iv) Investigators being permitted to lower the dose of treatment at signs of agitation;
A See response above regarding Pfizer and Glaxo SmithKline studies.
In the absence of administrative support to dig out specific records from these Prozac studies, I will email you the deposition of Dr Nancy Lord in Fentress v Lilly, who went through these Prozac records in detail and outlines a number of cases where data was miscoded, drug doses were lowered, concomitant medications were prescribed and patients who dropped out were not followed up.
The key pieces of this deposition start on page 75, with further key material around pages 98 et seq and 125 et seq. This deposition gives you patient numbers and identifiers to the precise pages which for the purposes of litigation were coded Pz 2618, 3797 etc, where Pz stands for Prozac.
I am attempting to track down Dr Lord’s further deposition in Berman v Lilly, which is even more precise regarding adverse event miscoding and the other matters you have asked for and will forward it just as soon as I have it.
Depending on how urgent this is, you may wish to contact Dr Nancy Zettler, the attorney in the Berman case, who is on Zet2400@aol.com. I can supply you with further contact details if needed.
v) Failure to follow-up patients who dropped out of the studies.
A This has been addressed to some extent above. But it is also worth adding specifically that this has been a feature of all trials of Zoloft/Lustral, Seroxat/Paxil and Prozac throughout, as far as I can make out.
The point has particular salience in the case of the trials in children, where both MHRA and FDA have repeatedly stated that no children have committed suicide, when in fact the correct statement would have been no children are known to have committed suicide, but it is also not known whether any in fact did commit suicide as a number of children who dropped out with serious adverse effects were lost to follow-up, and it is now clear for instance that the likelihood of suicidal acts during the period of withdrawal for instance from Seroxat is considerable.
Finally, I have a request to make in return. We have in fact ended up with an SSRI review committee, as I understand it, as a result of a process that began with my correspondence with you, and my concern that an ongoing failure to resolve the issues would do no one any good. My invitation to you initially was to show me where I am wrong.
Now I have little doubt but that MHRA will decide that it is not the brief of the SSRI committee to show Dr Healy where he is wrong, but I would respectfully suggest that a report stating there is no problem, which fails to address points such as those raised here, and which furthermore fails to make the data on which is based fully available, is not likely to settle matters.
I think you need to make some effort for instance to confirm
Whether suicidal acts on Prozac, Seroxat/Paxil and Zoloft/Lustral were coded in the way I outline above and in figures 1 & 2 or not,
Whether MHRA agrees with my interpretation of the meaning of confidence intervals outlined here, and if not what interpretation you would offer in lieu,
Whether MHRA reviewers have analysed the data on the complete set of symptoms displayed after Seroxat withdrawal rather than analysed the restricted set of data Glaxo SmithKline have pushed for or not,
Whether you in fact have managed to track down the Montgomery study in recurrent brief depression or not,
Whether you ever did get access to all documentation on the 1983 Hindmarch healthy volunteer study and whether you disagree with the assessment of the results made by the Pfizer monitor at the time.
Yours sincerely
Professor David Healy
====================================================
CLICK HERE FOR
INDEX OF HEALY CORRESPONDENCE WITH
THE MCA/MHRA