Prevention &
Treatment, Volume 5, Article 23, posted July 15, 2002
Copyright 2002 by the
American Psychological Association
The Emperor's New Drugs: An Analysis of Antidepressant Medication Data Submitted to the U.S. Food and Drug Administration
Irving Kirsch
University of Connecticut
Thomas J. Moore
The George Washington
University School of Public Health and Health Services
Alan Scoboria and Sarah S.
Nicholls
University of Connecticut
ABSTRACT
This article reports an analysis of the efficacy data submitted to the U.S. Food and Drug Administration for approval of the 6 most widely prescribed antidepressants approved between 1987 and 1999. Approximately 80% of the response to medication was duplicated in placebo control groups, and the mean difference between drug and placebo was approximately 2 points on the 17-item (50-point) and 21-item (62-point) Hamilton Depression Scale. Improvement at the highest doses of medication was not different from improvement at the lowest doses. The proportion of the drug response duplicated by placebo was significantly greater with observed cases (OC) data than with last observation carried forward (LOCF) data. If drug and placebo effects are additive, the pharmacological effects of antidepressants are clinically negligible. If they are not additive, alternative experimental designs are needed for the evaluation of antidepressants.
Keywords: drug efficacy, placebo, meta-analysis, depression
Correspondence concerning this article should be addressed
to to Irving Kirsch, Ph.D., Department of Psychology, University of
Connecticut , 406 Babbidge Road, U-20, Storrs, CT 06269-1020.
E-mail:
irving.kirsch@uconn.edu
Although antidepressant medication is widely regarded as efficacious, a recent meta-analysis of published clinical trials indicates that 75 percent of the response to antidepressants is duplicated by placebo (Kirsch & Sapirstein, 1998). These data have been challenged on a number of grounds, including the restriction of the analyses to patients who had completed the trials, the limited number of clinical trials assessed, the methodological characteristics of those trials, and the use of meta-analytic statistical procedures (Klein, 1998).
The present article reports analyses of a data set to which these objections do not apply, namely, the data submitted to the U.S. Food and Drug Administration (FDA) for approval of recent antidepressant medications. We analyzed the efficacy data submitted to the FDA for the six most widely prescribed antidepressants approved between 1987 and 1999 (RxList: The Internet Drug Index, 1999): fluoxetine (Prozac), paroxetine (Paxil), sertraline (Zoloft), venlafaxine (Effexor), nefazodone (Serzone), and citalopram (Celexa). These represent all but one of the selective serotonin reuptake inhibitors (SSRI) approved during the study period. The FDA data set includes analyses of data from all patients who attended at least one evaluation visit, even if they subsequently dropped out of the trial prematurely. Results are reported from all well controlled efficacy trials of the use of these medications for the treatment of depression. FDA medical and statistical reviewers had access to the raw data and evaluated the trials independently. The findings of the primary medical and statistical reviewers were verified by at least one other reviewer, and the analysis was also assessed by an independent advisory panel. More important, the FDA data constitute the basis on which these medications were approved. Approval of these medications implies that these particular data are strong enough and reliable enough to warrant approval. To the extent that these data are flawed, the medications should not have been approved.
Khan, Warner, and Brown (2000) recently reported the results of a concurrent analysis of the FDA database. Similar to the Kirsch and Sapirstein report, their analysis revealed that 76% of response to antidepressant was duplicated by placebo. In several respects, our analyses of the FDA data differ from, and supplement those, reported by Khan et al. First, although information on all efficacy trials for depression are included in the FDA database, mean change scores were not reported to the FDA for some trials on which a significant difference between drug and placebo was not obtained. Thus, the summary data reported by Khan et al. overestimate drug/placebo differences. In contrast, we provide an estimate of drug/placebo differences that is based on those medications for which for all clinical trials were reported, thus eliminating the bias due to the exclusion of trials least favorable to the medication.
Second, the means reported by Khan et al. (2000) were not adjusted for sample size. Thus, trials with small numbers of participants were given equal weight with the more reliable data from larger trials. In our analysis, mean scores were weighted by sample size, and summary statistics were calculated across medications for which full data were available.
Third, two methods of accounting for attrition were used in the data reported to the FDA: last observation carried forward (LOCF) and observed cases (OC). In LOCF analyses, when a patient drops out of a trial, the results of the last evaluation visit are carried forward as if the patient had continued to the completion of the trial without further change. In OC analyses, the results are reported only for those patients who are still participating at the end of the time period being assessed. Because patients who discontinue medication are regarded as treatment failures, LOCF analyses are widely considered to provide a more conservative test of drug effects, and the Khan et al. (2000) analysis was confined to those data. We used the FDA database to test this hypothesis empirically by comparing LOCF and OC data for all trials in which both were reported.
Finally, in many of the trials reported to the FDA, various fixed doses of the active medication were evaluated in separately randomized arms. Finding a dose-response relationship is one method of establishing the presence of true drug effects. Also, a dose-response relationship suggests that the drug effect may be underestimated in trials involving low dosages. Therefore, our analyses include a comparison of treatment effects at the lowest doses employed in fixed-dose trials with those at the highest doses.
Method
Using the Freedom of Information Act, we obtained the medical and statistical reviews of every placebo controlled clinical trial for depression reported to the FDA for initial approval of the six most widely used antidepressant drugs approved within the study period. We received information about 47 randomized placebo controlled short-term efficacy trials conducted for the six drugs in support of an approved indication of treatment of depression. The breakdown by efficacy trial was as follows: fluoxetine (5), paroxetine (16), sertraline (7), venlafaxine (6), nefadozone (8), and citalopram (5). Data on relapse prevention trials were not analyzed.
In order to generalize the findings of the clinical trial to a larger patient population, FDA reviewers sought a completion rate of 70% or better for these typically 6-week trials. Only 4 of 45 trials, however, reached this objective. Completion rates were not reported for two trials. Attrition rates were comparable between drug and placebo conditions. Of those trials for which these rates were reported, 60% of the placebo patients and 63% of the study drug patients completed a 4-, 5-, 6-, or 8-week trial. Thirty-three of 42 trials lasted 6 weeks, 6 trials lasted 4 weeks, 2 lasted 5 weeks, and 6 lasted 8 weeks. Patients were evaluated on a weekly basis. For the present meta-analysis, the data were taken from the last visit prior to trial termination.
Although the FDA approved the drugs for "the treatment of depression" not otherwise specified, all but one of the clinical trials were conducted on patients described as moderately to severely depressed (their mean baseline Hamilton Depression Scale [HAM-D] scores ranged from 21.0 to 29.7). One of the trials was conducted on patients with mild depression (mean baseline HAM-D score = 17.21). Thirty-nine of the 47 clinical trials focused on outpatients, 3 included both inpatients and outpatients, 3 were conducted with elderly patients (including one of the trials with both inpatients and outpatients), and 2 were conducted among patients hospitalized for severe depression. No trial was reported for the treatment of children or adolescents.
After 2 weeks, replacement of patients was allowed for those who investigators determined were not improving in three fluoxetine trials and in the three sertraline trials for which data were reported. The trials also included a 1- to 2-week placebo washout period, during which patients were given placebo. Those whose scores improved 20 percent or more were excluded from the study. The use of other psychoactive medication was reported in 25 trials. In most trials, a chloral hydrate sedative was permitted in doses ranging from 500 mg to 2000 mg per day. Other psychoactive medication was usually prohibited but still was reported as having been taken in several trials.
A shortcoming in the FDA data is the absence in many of the reports of reported standard deviations. This precludes direct calculation of effect sizes. Calculating effect sizes by dividing mean differences by standard deviations allows researchers to combine the results of trials on which different outcome measurement scales had been used. However, when the same scale is used across studies, it is possible to combine the results of the studies without first dividing them by the standard deviation of the scales (Hunter & Schmidt, 1990). The HAM-D was the primary endpoint for all of the reported trials in this analysis, thereby allowing direct comparisons of outcome data without conversion into conventional effect size (D) scores. The HAM-D is a widely used measure of depression, with interjudge reliability coefficients ranging from r = .84 to r = .90 (Hamilton, 1960).
For each clinical trial, we recorded the mean improvement in HAM-D scores in the drug and placebo groups. Next, improvement in the placebo group was divided by improvement in the drug group to provide an estimate of the degree of improvement in the drug-treated patients that was duplicated in the placebo group. Then, the mean of each of these trials, weighted for sample size, was calculated within each drug.
Results
Sample size and mean change on the HAM-D in drug and placebo conditions are presented in Table 1 for each of the 38 clinical trials on which LOCF data were reported.
| Table 1 | |||||
| Mean LOCF HAM-D Change in Drug and Placebo Conditions on Each Clinical Trial | |||||
|
| |||||
| Drug and study | Drug | Placebo | |||
|
|
|
||||
| Change | N | Change | N | ||
|
| |||||
| Fluoxetine | |||||
| 19 | -12.50 | 22 | -5.50 | 24 | |
| 25 | -7.20 | 18 | -8.80 | 24 | |
| 27 | -11.00 | 181 | -8.40 | 163 | |
| 62 (mild) | -5.89 | 299 | -5.82 | 56 | |
| 62 (moderate) | -8.82 | 297 | -5.69 | 48 | |
| Paroxetine | |||||
| 01-001 | -13.50 | 24 | -10.50 | 24 | |
| 02-001 | -12.30 | 51 | -6.81 | 53 | |
| 02-002 | -10.90 | 36 | -5.77 | 34 | |
| 02-003 | -9.73 | 33 | -7.15 | 33 | |
| 02-004 | -12.70 | 36 | -7.61 | 38 | |
| 03-001 | -10.80 | 40 | -4.70 | 38 | |
| 03-002 | -8.00 | 40 | -6.22 | 40 | |
| 03-003 | -9.90 | 41 | -10.00 | 42 | |
| 03-004 | -10.40 | 37 | -6.65 | 37 | |
| 03-005 | -10.00 | 40 | -4.07 | 42 | |
| 03-006 | -9.08 | 39 | -2.97 | 37 | |
| Par 09 | -9.14 | 403 | -8.23 | 51 | |
| Sertraline | |||||
| 103 | -9.92 | 261 | -7.60 | 86 | |
| 104 | -10.60 | 142 | -8.20 | 141 | |
| 315 | -8.90 | 76 | -7.80 | 73 | |
| Venlafaxine | |||||
| 203 | -11.20 | 231 | -6.70 | 92 | |
| 301 | -13.90 | 64 | -9.45 | 78 | |
| 302 | -11.90 | 65 | -8.88 | 75 | |
| 303 | -10.10 | 69 | -9.89 | 79 | |
| 313 | -11.00 | 227 | -9.49 | 75 | |
| 206 | -14.20 | 46 | -4.80 | 47 | |
| Nefazodone | |||||
| 03A0A-003 | -9.57 | 101 | -8.00 | 52 | |
| 03A0A-004A | -8.90 | 153 | -8.90 | 77 | |
| 03A0A-004B | -11.40 | 156 | -9.50 | 75 | |
| 030A2-0004 / 0005 | -10.00 | 74 | -9.84 | 70 | |
| 030A2-0007 | -12.30 | 175 | -9.80 | 47 | |
| CN104-002 | -10.80 | 57 | -8.20 | 57 | |
| CN104-005 | -12.00 | 86 | -8.00 | 90 | |
| CN104-006 | -10.00 | 80 | -8.90 | 78 | |
| Citalopram | |||||
| 85A | -8.78 | 82 | -6.63 | 87 | |
| 91206 | -9.95 | 521 | -8.32 | 129 | |
| 89303 | -11.76 | 134 | -10.24 | 66 | |
| 86141 | -6.26 | 98 | -4.74 | 51 | |
|
| |||||
Mean improvement (weighted for sample size) for each of the six
medications is presented in Table
2.
| Table 2 | |||||
| Mean Improvement (Weighted for Sample Size) in Drug and Placebo Conditions, and Proportion of the Drug Response That Was Duplicated in Placebo Groups for Each Antidepressant | |||||
|
| |||||
| Drug | K | N | Improvement | ||
|
| |||||
| Drug | Placebo | Propor- tion | |||
|
| |||||
| Fluoxetine | 5 | 1,132 | 8.30 | 7.34 | .89 |
| Paroxetine | 12 | 1,289 | 9.88 | 6.67 | .68 |
| Sertraline | 3 | 779 | 9.96 | 7.93 | .80 |
| Venlafaxine | 6 | 1,148 | 11.54 | 8.38 | .73 |
| Nefazodone | 8 | 1,428 | 10.71 | 8.87 | .83 |
| Citalopram | 4 | 1,168 | 9.69 | 7.71 | .80 |
|
| |||||
| Note. Data were not reported from four paroxetine trials, four sertraline trials, and one citalopram trial in which no significant differences were found. K = number of trials. | |||||
The 17-item version of the HAM-D was used in all trials of paroxetine, sertraline, nefazodone, and citalopram. The 21-item version was used in trials of fluoxetine and venlafaxine. One citalopram trial reported scores on both the 17-item scale and the 21-item scale, and another reported scores on the 17-item scale and a 24-item version of the scale. We used the 17-item scores for citalopram studies because this version of the scale was used in all of the clinical trials of that medication. Calculation of response to drug and placebo for the two studies using different forms of the scale reveals that the drug/placebo comparison is comparable, regardless of which scale is used.
Mean improvement scores were not reported in 9 of the 47 trials. Specifically, four paroxetine trials involving 165 participants, four sertraline trials involving 486 participants, and one citalopram trial involving 274 participants were reported as having failed to achieve a statistically significant drug effect, but the mean HAM-D scores were not reported. This represents 11% of the patients in paroxetine trials, 38% of the patients in sertraline trials, and 23% of the patients in citalopram trials. In each case, the statistical or medical reviewers stated that no drug effect was found.
Including data from paroxetine and sertraline trials in summary statistics would produce an inflated estimate of drug effects. Therefore, to obtain an unbiased estimate of drug and placebo effects across medications, we calculated weighted means of all medications for which data on all clinical trials were reported. This included the data for fluoxetine, venlafaxine, and nefadozone. The weighted mean difference between the drug and placebo groups across these three medications was 1.80 points on the HAM-D, and 82% of the drug response was duplicated by the placebo response. A t-test, weighted for sample size, indicated that the drug/placebo difference was statistically significant, t(18) = 5.01, p < .001.
On most of the clinical trials, medication dose was titrated individually for each patient within a specified range. However, in 12 trials involving 1,942 patients, various fixed doses of a medication were evaluated in separately randomized arms. It is possible that some of the doses used in these trials were subclinical. If this is the case, inclusion of these data could result in an underestimate of the drug effect. To test this possibility, we compared LOCF data at the lowest and highest doses reported in each study. Across these 12 trials, mean improvement (weighted for sample size) was 9.57 points on the HAM-D at the lowest dose evaluated and 9.97 at the highest dose. This difference between high and low doses of antidepressant medication was not statistically significant.
Finally, we tested the hypothesis that LOCF analyses provide more conservative tests of drug effects than do OC analyses. LOCF means were reported for all 38 of the 46 trials in which means of any kind were reported. OC means were reported for 27 of these 38 trials. In 22 trials, the difference between drug and placebo group was not statistically significant with either LOCF or OC measures. In 12 trials, the difference was statistically significant with both measures. In 8 trials, the difference was significant with LOCF but not with OC, and 4 trials were reported to have shown no difference between drug and placebo without specifying an attrition rule. For the 27 trials for which both sets of means were reported, correlated t-tests indicated that mean improvement scores were significantly greater with OC data than with LOCF data for both drug, t(26) = 12.46, p < .001, and placebo, t(26) = 10.56, p < .001, as was the proportion of the drug response duplicated by placebo, t(26) = 3.36, p < .01. In the LOCF data, 79% of the drug response was duplicated in the placebo groups; in the OC data, 85% of the drug response was duplicated by placebo. Thus, LOCF analyses indicate a greater drug/placebo difference than do OC analyses.
Discussion
In clinical trials, the effect of the active drug is assumed to be the difference between the drug response and the placebo response. Thus, the FDA clinical trials data indicate that 18% of the drug response is due to the pharmacological effects of the medication. This is based on LOCF data, in which the drug effect was significantly stronger than in OC data, and it is obtained after those who show the greatest response to placebo are excluded from the study. Overall, the drug/placebo difference was less than 2 points on the HAM-D, a highly reliable physician-rated scale that has been reported to be more sensitive than patient-rated scales to drug/placebo differences (Murray, 1989). The range was from a 3-point drug/placebo difference for venlafaxine to a 1-point difference for fluoxetine, both of which were on the 21-item (64-point) version of the scale. As intimated in FDA memoranda (Laughren, 1998; Leber, 1998), the clinical significance of these differences is questionable.
The proportion of the drug response duplicated in placebo groups is greater in the FDA clinical trials data than in previous meta-analyses (Khan et al., 2000; Kirsch & Sapirstein, 1998). The differences may be due to two factors: publication bias and missing data. Publication bias is avoided in the FDA data by the requirement that the results of all trials for an indication be reported. Calculating summary statistics only for medications for which means on all trials were reported circumvented the missing data problem.
Of the two widely used methods of coping with attrition in clinical trials, LOCF analyses are considered the more stringent. The FDA data set calls this assumption into question. The proportion of the drug effect duplicated by placebo was significantly larger in the OC data set than in the corresponding LOCF data set. In addition, the degrees of freedom are necessarily larger in LOCF analyses, thereby making it more likely that a mean difference will be statistically significant. In the 47 clinical trials obtained from the FDA, there were no reported instances in which OC data yielded significant differences that were not detected in LOCF analyses. However, in 8 trials, LOCF data yielded significant differences that were not detected when OC data were analyzed. These data indicate that, compared with LOCF analyses, OC analyses provide more conservative tests of drug/placebo differences.
Although mean differences were small, most of them favored the active drug, and overall, the difference was statistically significant. There were only 4 trials in which mean improvement scores in the placebo condition were equal to or higher than those in the drug condition, and in no case was placebo significantly more effective than active drug. This may indicate a small but significant drug effect. However, it is also possible that this difference between drug and placebo is an enhanced placebo effect due to the breaking of blind. Antidepressant clinical trial data indicate that the ability of patients and doctors to deduce whether they have been assigned to the drug or placebo condition exceeds chance levels (Rabkin et al., 1986), possibly because of the greater occurrence of side effects in the drug condition. Knowing that one has been randomized to the active drug condition is likely to enhance the placebo effect, whereas knowledge of assignment to the placebo group ought to decrease its effect (Fisher & Greenberg, 1993). Enhanced drug effects due to breaking blind in clinical trials may be small, but evaluation of the FDA database indicates that the drug/placebo difference is also very small, amounting to about 2 points on the HAM-D.
Although our data suggest that the effect of antidepressant drugs are very small and of questionable clinical significance, this conclusion rests on the assumption that drug effects and placebo effects are additive. However, it is also possible that antidepressant drug and placebo effects are not additive and that the true drug effect is greater than the drug/placebo difference. Clinical trials are based on the assumption of additivity (Kirsch, 2000). That is, the drug is deemed effective only if the response to it is significantly greater than the response to placebo, and the magnitude of the drug effect is assumed to be the difference between the response to drug and the placebo. However, drug and placebo responses are not always additive. Alcohol and stimulant drugs, for example, produce at least some drug and placebo effects that are not additive. Placebo alcohol produces effects that are not observed when alcohol is administered surreptitiously, and alcohol produces effects that are not duplicated by placebo alcohol (Hull & Bond, 1986). The placebo and pharmacological effects of caffeine are additive for feelings of alertness but not for feelings of tension (Kirsch & Rosadino, 1993), and similarly mixed results have been reported for other stimulants (Lyerly, Ross, Krugman, & Clyde, 1964; Ross, Krugman, Lyerly, & Clyde, 1962).
If antidepressant drug effects and antidepressant placebo effects are not additive, the ameliorating effects of antidepressants might be obtained even if patients did not know the drug was being administered. If that is the case, then antidepressant drugs have substantial pharmacologic effects that are duplicated or masked by placebo. In this case, conventional clinical trials are inappropriate for testing the effects of these drugs, as they may result in the rejection of effective medications. Conversely, if drug and placebo effects of antidepressant medication are additive, then the data clearly show that those effects are small, at best, and of questionable clinical efficacy. Finally, it is conceivable that the effects are partially additive, with the true drug effect being somewhere in between these extremes. The problem is that we do not know which of these models is most accurate because the assumption of additivity has never been tested with antidepressant mediation.
One method of testing the additivity is the use of the balanced placebo design (Marlatt & Rohsenow, 1980). In this design, informed consent is first obtained for a study in which active drug or placebo will be administered. Half of the participants are told they are receiving active drug and half are led to believe they are not. In fact, half of the participants are given an active drug and half are not. Thus, half of the participants are misinformed about what they will receive and are debriefed after participation in the trial. As shown in Figure 1, there are four cells in the balanced placebo design.
Depending on assignment, participants are (a) told they are getting the
drug and do in fact receive it, (b) told they are getting drug but in fact
receive placebo, (c) told they are getting placebo but in fact receive
drug, and (d) told they are getting placebo and in fact receive placebo.
This permits independent and combined assessment of drug and placebo
effects. This design has been used with healthy volunteers and has provided
interesting data on the additive and nonadditive effects of alcohol (Hull
& Bond, 1986) and caffeine (Kirsch
& Rosadino, 1993). It has not been used in clinical trials, in
which its use might pose a more difficult ethical problem because of the
temporary deception that is involved. However, there is also an ethical
risk involved in not assessing the additivity assumption underlying
clinical trials. If that assumption is unwarranted, effective medications
may be rejected because their effects are masked by placebo effects.
Conversely, if the assumption is warranted, then current antidepressants
may be little more than active placebos. Thus, some means of assessing the
additivity hypothesis is a crucial task. Without the assumption of additivity, the FDA data do not allow one to
determine the effectiveness of antidepressant medication. That is, it is
not possible to determine the degree to which the antidepressant response
is a drug effect and the degree to which it is a placebo effect. If one
does make the assumption that the drug effect is the difference between
the drug response and the placebo response, then it is very small and of
questionable clinical value. By far, the greatest part of the change is
also observed among patients treated with inert placebo. The active agent
enhances this effect, but to a degree, that may be clinically
meaningless. These data raise questions about the criteria used by the FDA in
approving antidepressant medications. The FDA required positive findings
from at least two controlled clinical trials, but the total number of
trials can vary. Positive findings consist of statistically significant
drug/placebo differences. The clinical significance of these differences
is not considered. The problems associated with these criteria are illustrated in a
memorandum from the director of the FDA Division of Neuropharmacological
Drug Products (DNDP; Leber,
1998) on the approvable action on Celexa (citalopram) for the
management of depression. Two controlled efficacy trials showed
significant drug/placebo differences. Three others "failed to provide
results confirming the positive findings" (Leber,
1998, p.6).1
This led to the conclusion that "there is clear evidence from more than
one adequate and well controlled clinical investigation that citalopram
exerts an antidepressant effect. The size of that effect, and more
importantly, the clinical value of that effect, is not something that can
be validly measured, at least not in the kind of experiments conducted.
Accordingly, substantial evidence in the present case, as it has in all
other evaluations of antidepressant effectiveness, speaks to proof in
principle [emphasis added] of a product's effectiveness" (Leber,
1998, p. 7). Similarly, the DNDP team leader for psychiatric drug products
commented, "While it is difficult to judge the clinical significance of
this difference, similar findings for other SSRIs and other recently
approved antidepressants have been considered sufficient to support the
approvals of those other products" (Laughren,
1998, p. 6). Laughren noted that "while the reasons for negative
outcomes for [these studies] are unknown," about 25% of the patients in
one of the failed studies did not meet criteria for major depression, and
in the other two, "there was a substantial placebo response, making it
difficult to distinguish drug from placebo" (Laughren,
1998, p. 4). On the basis of these concerns, he concluded, "I feel
there were sufficient reasons to speculate about the negative outcomes
and, therefore, not count these studies against citalopram" (Laughren,
1998, p. 6). To summarize, the data submitted to the FDA reveal a small but
significant difference between antidepressant drug and inert placebo. This
difference may be a true pharmacological effect, or it may be an artifact
associated with the breaking of blind by clinical trial patients and the
psychiatrists who are rating the severity of their conditions. Further
research is needed to determine which of these is the case. In any case, the difference is relatively small (about 2 points on the
HAM-D), and its clinical significance is dubious. Research is therefore
needed to assess the additivity of antidepressant drug and placebo
effects. If there is a powerful antidepressant effect, then it is being
masked by a nonadditive placebo effect, in which case current clinical
trial methodology may be inappropriate for evaluating these medications,
and alternate methodology need to be developed. Conversely, if the drug
effect is as small as it appears when drug/placebo differences are
estimated, then there may be little justification for the clinical use of
these medications. The problem, then, would be to find an alternative, as
the clinical response to both drug and placebo is substantial. Placebo
treatment has the advantage of eliciting fewer side effects. However, the
deception that is inherent in clinical administration of placebos inhibits
their use. Thus, the development of nondeceptive methods of eliciting the
placebo effect would be of great importance. Fisher, S., & Greenberg, R. P. (1993). How sound is the
double-blind design for evaluating psychotropic drugs. Journal of
Nervous and Mental Disease, 181, 345-350. Hamilton, M. A. (1960). A rating scale for depression. Journal of
Neurology, Neurosurgery, and Psychiatry, 23, 56-61. Hull, J. G., & Bond, C. F. (1986). Social and behavioral
consequences of alcohol consumption and expectancy: A meta-analysis.
Psychological Bulletin, 99, 347 360. Hunter, J. E., & Schmidt, F. L. (1990). Methods of
meta-analysis: Correcting error and bias in research findings. Newbury
Park, CA: Sage. Khan, A., Warner, H. A., & Brown, W. A. (2000). Symptom reduction
and suicide risk in patients treated with placebo in antidepressant
clinical trials: An analysis of the Food and Drug Administration database.
Archives of General Psychiatry 57, 311-317. Kirsch, I. (2000). Are drug and placebo effects in depression additive?
Biological Psychiatry 47, 733-73. Kirsch, I., & Rosadino, M. J. (1993). Do double-blind studies with
informed consent yield externally valid results? An empirical test.
Psychopharmacology, 110, 437-442. Kirsch, I., & Sapirstein, G. (1998). Listening to Prozac but
hearing placebo: A meta analysis of antidepressant medication.
Prevention & Treatment, 1, Article 0002a. Available on the
World Wide Web: http://www.journals.apa.org/prevention/volume1/pre0010002a.html Klein, D. F. (1998). Listening to meta-analysis but hearing bias.
Prevention & Treatment, 1, Article 0006c. Available on the
World Wide Web: http://www.journals.apa.org/prevention/volume1/pre0010006c.html Laughren, T. P. (1998, March 26). Recommendation for approvable
action for Celexa (citalopram) for the treatment of depression.
Memoradum: Department of Health and Human Services, Public Health Service,
Food and Drug Administration, Center for Drug Evaluation and Research,
Washington, DC. Leber, P. (1998, May 4). Approvable action on Forrest Laboratories,
Inc. NDA 20-822 Celexa (citalopram HBr) for the management of
depression. Memoradum: Department of Health and Human Services, Public
Health Service, Food and Drug Administration, Center for Drug Evaluation
and Research, Washington, DC. Lyerly, S. B., Ross, S., Krugman, A. D., & Clyde, D. J. (1964).
Drugs and placebos: The effects of instructions upon performance and mood
under amphetamine sulphate and chloral hydrate. Journal of Abnormal and
Social Psychology, 68, 321 327. Marlatt, G. A., & Rohsenow, D. J. (1980). Cognitive processes in
alcohol use: Expectancy and the balanced placebo design. In N. K. Mello
(Ed.), Advances in substance abuse: Behavioral and Biological
Research, (pp. 159 199). Greenwich, CT: JAI Press. Murray, E. J. (1989). Measurement issues in the evaluation of
pharmacological therapy. In S. Fisher & R. P.Greenberg (Eds), The
limits of biological treatments for psychological distress: Comparisons
with psychotherapy and placebo (pp. 39-67). Hillsdale, NJ:
Erlbaum. Rabkin, J.G., Markowitz, J. S., Stewart, J. W., McGrath, P. J.,
Harrison, W., Quitkin, F. J., & Klein, D. F. (1986) How blind is
blind? Assessment of patient and doctor medication guesses in a
placebo-controlled trial of imipramine and phenelzine. Psychiatry
Research, 19, 75-86. Ross, S., Krugman, A. D., Lyerly, S. B., & Clyde, D. J. (1962).
Drugs and placebos: A model design. Psychological Reports, 10, 383
392. RxList: The Internet Drug Index. (1999). The top 200 prescriptions
for 1999 by number of U.S. prescriptions dispensed. Retrieved November
19, 2001, from http://www.rxlist.com/99top.htm 1Data on two maintenance studies were also reported by the
manufacturer of Celexa. In these relapse prevention trials, participants
who had responded to citalopram were ramdomized to drug or placebo. HAM-D
scores did not distinguish between drug and placebo in one of these trials
and were not assessed in the other. The primary outcome in these studies
was time to relapse (Laughren,
1998). Mean time to relapse was 21 weeks for citalopram versus 18
weeks for placebo in one of these studies and was not reported in the
other.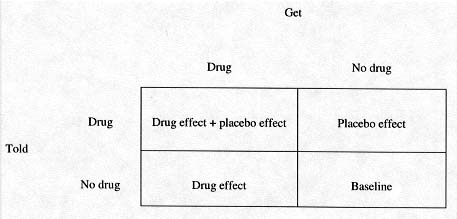
Figure 1. The
balanced placebo design.
References
Footnote